 The original of this image can be found
in: http://hcs.harvard.edu/~husn/BRAIN/vol7-spring2000/neuroimmuno.htm.
The original of this image can be found
in: http://hcs.harvard.edu/~husn/BRAIN/vol7-spring2000/neuroimmuno.htm.Mammalian Toxicology, Session 5
Cell Death: Necrosis, Apoptosis; C 3, 10, 18, 20; Neural and Immune Function Overview; C 3, 12, 16
It is anticipated that all groups formed last week will have been in active communication this past week and have approached some conclusions on what project topic they will be attacking over the course of the next few weeks. Note that the topic choice along with a brief argument of why it is important needs to be posted by e-mail or on the Prometheus site by the end of this week. Please make every effort to complete that assignment. Also note that I will continue to post some additional discussion questions to keep people thinking about the reading assignments over the coming weeks. Don't forget to spend a little time on these even as you work on the projects.
Also note the following site as a good prospect for reviewing basic material covered to date:
http://www.biology.arizona.edu/chh/problem_sets/toxicology/toxicology.html
Cell Death
Some of the material here was covered in connection with the material on Cell Cycle Controls presented earlier. I will refer you back to that material for some of the coverage and simply expand a bit on those comments.
Toxicological insult to an organism may involve temporary disruption of function or permanent dysfunctions up to and including organismal death. Dysfunctions may be metabolic, but they often involve actual physical damage to tissues. ndeed, this is how many of the initial evaluations of toxic response are visualized and/or quantified. Organs change in size, morphology, or cellular and acellular composition, and tissues display deviations from normal form upon gross or microscopic histological (examination of tissues, usually after chemical fixation, sectioning, and staining) or cytological (examination of cell suspensions, often as live or minimally manipulated preparations) evaluation. Those changes in tissues often involve the excessive death of segments of the usual cell population. Note I said excessive. Most adult tissues undergo cyclical renewal so the cells contained are replaced at a rate balanced by the normal rate of cell death, i.e., the kinetics of cell renewal and removal are balanced at equilibrium. Even during growth and development when replacement often exceeds cell death, the system deviates in a very controlled manner from steady state equilibrium. But toxic insult can change that balance. Here we consider excessive cell death. When carcinogenesis is addressed, excessive cell replacement and growth are involved.
But what is cell death? And how does it occur?
Two paths to cell death are well defined in the literature: necrosis and apoptosis. Necrosis is usually equated with rapid or traumatic cellular rupture and is usually associated with inflammation and immune activation. Apoptosis, on the other hand, is a slower, ordered process in which the affected cell gradually undergoes autolysis of its intracellular components and ultimately vesiculates and is removed by adjacent non-immune cells or by resident reticuloendothelial components with minimal or no recruitment of more remote immune cells. Toxicants or toxins can selectively act to initiate one or the other of these forms of cell death. The higher the elevation of cell death rate, by either mechanism, above the normally encountered rate, the more compromised the target tissue becomes and the more likely this morbidity is to be detrimental to the intact organism. In the central nervous system, where neuronal replacement is very low or nonexistant, or in the crystalline lens of the eye or among the oocyte complement of the mammalian ovary, where cell division and differentiation are completed soon after birth, such damage will be permanent regardless of the mode of cell death. In contrast, in tissues such as the seminiferous epithelium, the bone marrow, the epidermis, or the intestinal epithelium which experience very rapid cellular replacement, toxic insults involving cellular death may often be temporary unless the die-offs are massive and trigger secondary problems. These generalizations will definitely hold for the induction of apoptotic cell death. In the case of necrosis, however, the rapid and immediate involvement of the immune system will often trigger reactions that are themselves potentially deleterious (more below).
While the cellular components of bone and most other tissues including excitable cardiac muscle and smooth muscles follow the behaviors noted above, skeletal muscle presents an interesting situation. Differentiated striated muscle is syncytial with many nuclei in each large cell. Now we know that muscles can get larger or smaller depending on nutrient input, health, and level of physical activity. Similar changes will also occur with toxic insult. But how is that occurring? Clearly, trauma will cause necrosis and loss of entire muscle cells. But will toxicants or inactivity cause selective multinucleate cell death via apoptosis? Or will segments of fibers shrink and lose nuclei in a random, stochastic manner and regrow by mitosis of remaining nuclei followed by a lack of diakinesis (cell division)? To my knowledge, this form of tissue involution and replacement or cell death is still mechanistically unresolved. Any volunteers to unravel the problem?
C&D lay out the three basic insults that lead to cell death: ATP depletion, sustained rises in intracellular calcium ion, and overproduction of reactive oxygen or nitrogen species such as superoxide or nitrous oxide radicals. If these occur in a concerted manner there is an irreversible insult leading to necrosis. If, on the other hand the insult only involves one or the other of these problems or they arise in a gradual manner that can be regulated by the normal regulatory paths and repair functions within the insulted cell, then cell death may be avoided or at least an ordered progression of cell death events can occur via the apoptotic pathways.
Necrosis
The "overt killing of cells... rapid cell death usually due to sudden breach of the cell membrane or rapid loss of membrane ion gradients or osmotic gradients followed by infiltration with inflammatory cells such as lymphocytes and monocytes" as I put it in an earlier session. Or as defined in Stedman's Medical Dictionary, 24th Edition (Williams & Wilkins: Baltimore, MD, 1983), page 929: "The pathologic death of one or more cells, or of a portion of tissue or organ, resulting from irreversible damage; the most frequent visible alterations are nuclear: pyknosis, i.e., shrunken and abnormally dark basophilic staining; karyolysis, i.e., swollen and abnormally pale basophilic staining; or karyorrhexis, i.e., rupture and fragmentation of the nucleus. After such changes, the outlines of individual cells are indistinct, and affected cells may become merged, sometimes forming a focus of coarsely granular, amorphous, or hyaline material. The earliest irreversible changes are mitochondrial, consisting of swelling and granular calcium deposits seen by electron microscopy."
Note in the medical evaluation of cell death which is largely based on histological observations, the importance of mitochondrial changes is already underlined. ATP production and calcium ion sequestration are both major features of mitochondrial function. Disrupting either of these leads to a cascade or problems within cells. Blocking ATP production driven by the mitochondrial electron transport chain and the proton gradient-driven ADP kinase deprives cells of the main energy source needed to power the ion pumps in both the intracellular organelles and the cell membrane. Those pumps maintain the chemical and electrical gradients necessary to regulate cellular functions and maintain intracellular homeostasis. When they fail, control of osmotic regulation is lost and cells will literally burst as potassium, calcium, and water rush into the cell. But their failure also allows loss of compartmentation within the cell so that enzymes such as the proteases, lipases, nucleases, and glycolases that reside in lysosomes or that exist in inactivated states in the cell cytoplasm may become activated. Reactive oxygen and nitrogen species disrupt electron transport in several cytochromes and alter the redox states of metalloproteins leading to establishment of spirals of reactive oxygen and nitrogen species production or of lipolysis. Interestingly, the rapid chain of events leading to cell necrosis leads to release of intracellular materials (cytochrome c?, small molecular mediators?) that act as chemoattractants for phagocytic immune cells such as monocytes and macrophages. These then generate the inflammatory response typical following necrotic cell death.
While C&D classify the cell deaths associated with direct cell membrane disruption by detergents or solvents as undefined, it is difficult to envision how these might occur by a pathway grossly different from necrosis given the importance of membrane breaching in the pathway leading to necrosis.
Apoptosis
Stedman (circa 1983) offers this definition on page 99: "Cell deletion by fragmentation into membrane-bound particles which are phagocytosed by other cells." Note that by 2000 this had not only expanded to a description of programmed cell death in most discussions of the topic, but it had become an area of very active scientific activity. Some of this is captured or accessible via the material from biomedical companies like Boehringer-Mannheim,
http://biochem.boehringer-mannheim.com/prod_inf/manuals/cell_man/cell_toc.html
or via compilations such as the Virtual Library of Cell Biology: http://vlib.org/Science/Cell_Biology/apoptosis.shtml.
But additional aspects are covered by The Cell Death Society and their journal Apoptosis:
http://www.celldeath-apoptosis.org/.
Key in description of apoptosis is the orderliness of the process and it's potential reversibility. As stated last session apoptosis "is often triggered by p53 activation or by the presence of alternative triggering paths like Fas/Fas ligand interactions. During toxic insults the path is often activated by the intracellular production of oxidative products such as peroxide or superoxide. Many indications seem to point to the production of damage to the mitochondrial membrane and intra- as well as extracellular release of cytochrome c as a key element in activating transcription and translation of genes associated with the apoptotic cascade (e.g., caspases, Bax, Bcl) that leads to intracellular proteolysis, nucleolysis, organellar breakdown, and finally cellular disruption by osmotic pressure and debris removal by adjacent cells." Toxicants would not trigger frequently the ligand mediated pathways to apoptosis except by indirect routes such as depletion of some necessary growth promoting factor via blockade of its uptake or by activation of immune pathways leading to identification of the tissue as "non-self." Rather, toxic insults would often tend to activate the intracellular triggers to the caspase cascade and the expression of cell death proteins. Ultimately these enzymatic activity changes within the cell cause loss of organellar content and integrity with intracellular vacuolization, including shrinking and/or fragmentation of the cell nucleus. But they only move toward the same kinds of terminal events seen in necrosis, except the release of inflammatory cell chemotactic factors, at the very late stages of the process. Rather than "exploding," these cells involute.
And early in apoptosis, there is the potential for cellular rescue via the actions of extracellular factors activating paths that stimulate expression of Bcl or related proteins that inhibit the caspase cascade. So activation of the cell death path is at least initially reversible, something not possible in a necrotic insult. Metabolic breakdown of reactive species or production of sufficient quantities of ATP to allow reversal of losses in intracellular calcium ion control can allow cells to avoid death if apoptosis is involved.
Neural Function Overview
C&D coverage of neural function is limited to a very general discussion of toxic impacts on nervous tissue. That in the peripheral nervous system (PNS) being repairable if the insult is limited to impacts on the peripheral axonal projections and their intracellular machinery while that in the central nervous system (CNS) is essentially irreversible for any insult. In both cases nerve loss in the adult mammal cannot be repaired in the vast majority of instances because of the lack of available stem cells in either the PNS or the CNS. Necrotic or apoptotic impacts on either cell population result in nervous system deficits. Interestingly, the unusual geometry and intracellular makeup of nerves possessing long or extensive axonal projections allows these cells to lose a part or most of an axon without triggering paths to cell death. This, in turn, opens the way for regrowth of axonal projections in the PNS. In the CNS, on the other hand, the lack of production of nerve growth factor or similar growth promotors by glial or other associated cells prevents axonal regrowth. As a result, toxicants that target CNS axonal components likely will have permanent impacts on the affected organism while toxicants that target PNS axonal components may have temporary impacts.
The chemical nature of neurotransmitters and their unique metabolic, secretion, and reuptake mechanisms in the synaptic cleft and neuromuscular junctions also provide targets for toxicant actions. Chemicals that act as agonists or antagonists for the many neurotransmitters (both stimulatory, e.g., epinephrine, acetyl choline, glutamate, and inhibitory, e.g., gamma amino butyric acid, glycine) can interfere with normal neural communication by several paths. Agonists may cause persistent stimulation of post-synaptic receptors and therefore trigger downstream neurons to undergo continuous or repeated depolarizations. They can block metabolic enzymes and prevent the natural transmitter from breaking down after release into the synaptic cleft thereby resulting in a supra-physiological buildup of those signals. Or, they can block reuptake of the natural ligand and cause persistent activation of downstream neurons. Antagonists, on the other hand, can block post-synaptic receptors and prevent normal transmission from occurring. Agents like ether may also alter synaptic membrane fluidity and change the kinetics of signal transmission and/or ion channel dynamics. These insults do not have to be irreversible or lead to cell death. They form the basis for the actions of most pharmaceuticals that alter neuronal or neuromuscular function including anesthetics, analgesics, and psychoactive agents such as neuroleptics or antipsychotics. It is only when the pharmacological effects are exaggerated by elevated available dosages or by exposure to similarly neuroactive compounds that neurotoxic impacts become apparent.
The nature of the blood-brain barrier needs a bit of clarification as there is at least one misstatement concerning its nature in C&D. This barrier is formed as noted by the singular properties of the vascular system throughout most of the brain combined with the tight coverage of neurons by glial cells of the CNS. The vascular endothelial cells lack the normal fenestrations (seive-like, membrane-bound perforations that extend through the cell membrane and cytoplasm rather like donut holes) that occur near the tails of most endothelial cells elsewhere in the body. The endothelia lining most of the cerebral vasculature also is more tightly arranged than elsewhere. Moreover, these cells produce tight junctional complexes that seal off the vascular lumen from the extravascular, brain compartment. The lack of intracellular spaces caused by the tight junctional complexes combined with the lack of fenestrations normally prevent immune cells, proteins, and hydrophilic compounds from gaining access to the neural tissues. This is reinforced by the tight packing of glial projects around the neurons. With this arrangement, even lipophilic compounds must diffuse across at least four cell membranes before they gain access to neuronal tissues. Proteins or charged compounds have to be transported via specific transport systems while immune cells only gain access when the barriers are temporarily modified by elevations in certain signaling compounds such as interleukins. Note that the use of tight junctional complexes between cells is not unique to the cerebral vascular endothelium. It occurs in any tissue or organ in which there is a need to segregate the contents of a lumenal space from surrounding tissues. Thus, Sertoli cells in the seminiferous tubules seal off the tubule lumen from extra-tubular space (and help form the blood-testis barrier), gastric cells are joined by tight junctions to prevent movement of gastric contents through the stomach wall into the peritoneum, thyroid follicular cells seal off the thyroid follicle lumen from the extra-follicular tissues, etc. The lack of endothelial fenestrations and the presence of the glial barrier is unique. But do understand that these endothelial specializations are missing from certain portions of the brain including the portal vasculature of the hypothalamus (in fact the hypothalamic-pituitary axis could not function if the blood-brain barrier was intact in this region). So this barrier protects most, but not all of the CNS and does not apply in the PNS where isolation is only via the myelin coating afforded by Schwann cells.
The links below address basic neuroanatomy or neurobiology:
http://www.med.harvard.edu/AANLIB/home.html
http://directory.google.com/Top/Science/Biology/Neurobiology/
The following links provide further information on neurotoxicology:
http://www.emedicine.com/neuro/NEUROTOXICOLOGY.htm
Immune Function Overview
The coverage of immune function and toxic responses of the immune system in C&D is good and should be thoroughly read. Several very well illustrated books on basic immunology and immunoanatomy by David Male are also to be recommended as well as the following links:
http://www.immunologylink.com/
http://gsbs.utmb.edu/microbook/ch001a.htm
http://www.biology.arizona.edu/immunology/immunology.html
http://www.bio.davidson.edu/courses/Immunology/Bio307.html
http://www.med.sc.edu:85/book/immunol-sta.htm
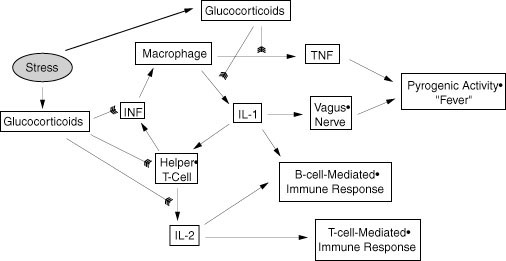
In all these coverages the duality of the immune system is described: innate versus acquired, cellular versus humoral, nonspecific versus specific. It is also made clear that the cells of the immune system form a complex interactive network that uses all forms of chemical signaling (endocrine, autocrine, paracrine, and juxtacrine) in carrying out their tasks. An extensive list of proteinaceous interleukins, cytokines, and growth factors is listed in C&D (Table 12-5, page 426-428). In addition we should add the eicosanoid leukotrienes as paracrine modulators, the glucocortical steroids including cortisol, corticosterone, 11-deoxycorticosterone, and the progestational steroid progesterone as mainly immunosuppressive lipophyllic hormones, and ACTH and CRH as indirect regulators of immune function. The inter-relationships among the immune, endocrine, and nervous system are becoming clearer every day as are the relationships between mental/emotional status and physical health status, particularly as overseen by immune surveillance. We will later spend a bit more time on stress and its impacts on toxic insults. The interplay among the immune and communications systems plays a key role in both the normal and abnormal stress responses.
Here I just want to underline how the immune system is suppressed by the actions of glucocorticoids at the level of protein expression in immune cells (especially T and B lymphocytes). In the extreme case, as in the developing thymus, this will also trigger apoptosis in the lymphoid cells. Note that progesterone can bind to the glucocorticoid receptors when present at high concentrations such as occur during pregnancy, or during the latter half of the ovarian cycle (the luteal phase). This provides part of the explanation for maternal immune suppression during pregnancy and for the mild hyperthermia that occurs just at the beginning of the luteal phase of the ovarian cycle. But this is only one of the interactive linkages. Interleukins 1 and 2 are capable of increasing CRH production in the hypothalamus. This, in turn, increases ACTH production by pituitary corticotropes and thereby stimulates production of glucocorticoids in the adrenal cortex. Thus, there is a negative feedback control loop that limits the extent of immune response under normal homeostatic conditions. This loop affects both the innate and acquired immune system because it impacts both T and B cell lineages.
The original of this image can be found
in: http://hcs.harvard.edu/~husn/BRAIN/vol7-spring2000/neuroimmuno.htm.
Other major points concerning immunotoxicity relate to the sensitivity of the immune system to potential insult due to the necessary DNA modifications that go on during immune cell maturation and to the amount of apoptosis that normally occurs during immune system maturation. Both T and B cells undergo somatic DNA rearrangement in the course of clonal expansion and maturation in response to antigen interaction, antigen presentation or indirect activation. This means they must undergo a cleavage and rearrangement of part of the genome very much akin to what happens during meiosis in germ cells. The cellular machinery involved resembles that involved in DNA repair and has the same potential for errors leading to cell death or neoplasia. Thus, these cells are more sensitive than most somatic cells to possible toxic insults involving DNA synthesis and repair pathways. In addition, clonal expansion is a necessary component of the amplification of the immune response and the production of immune memory. Again, in any process involving rapid cellular division there are many opportunities for errors in DNA replication to occur and ultimately lead to cell death, mutation, and/or neoplasia if check points are ignored during the mitotic process. Additionally, during immune system maturation suppression of anti-self directed immune cells is accomplished by elimination of those cells via Fas/Fas ligand mediated apoptotic cell death. Apoptosis occurs subsequent to activation of the cells interaction with self antigens or with phagocytic cells that present portions of self-antigens. Toxins that impact the apoptotic process may interfere with normal immune function via elimination of too few or too many lymphocytic cell lineages.
The following link describes the complex process of somatic gene rearrangement as it takes place in normal cells and demonstrates the numerous points at which B and T cells and their progenitors can succumb to toxic insults:
http://webmed.unipv.it/immunology/ b&tcelldev.html
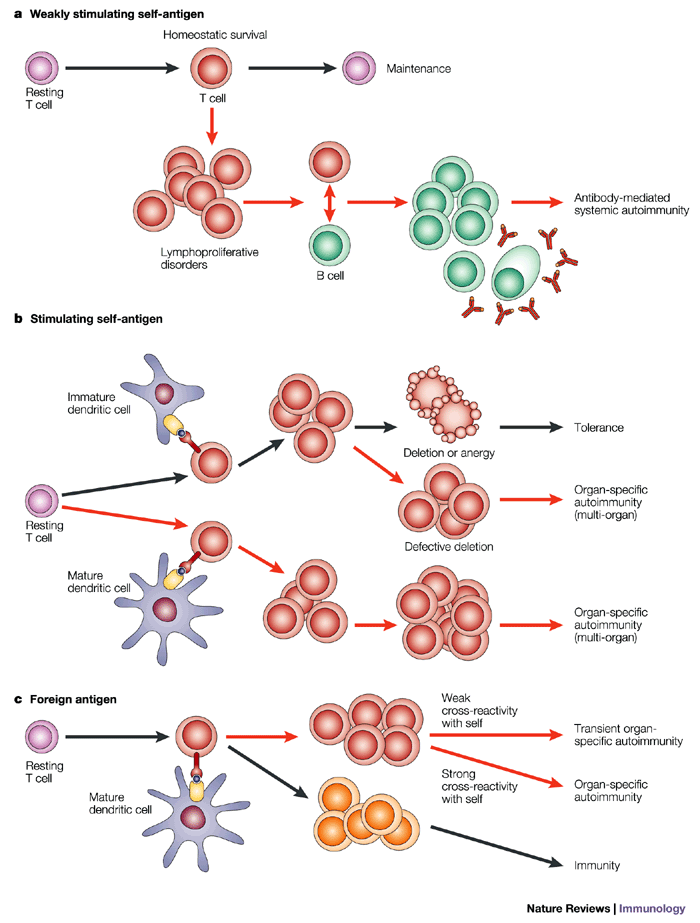
Obviously, any exogenous compound that can mimic a non-self antigen has the potential to trigger an immune reaction in a previously exposed individual. Alternatively, it can block a response by the natural antigen when they are simultaneously present. More likely, however, are the instances in which toxicants alter self molecules in such a way that they are recognized by the immune system as non-self. This will result either in activation of a cellular immune reaction causing damage to self tissue and/or to the production of antibodies. These antibodies may be directed either against the modified self antigen that will subsequently cross-react with and be targeted by the immunoglobulins or T-cell receptors and be selected and amplified, or they may be directed against self antigens that are normally not exposed to immune cells, e.g., nuclear proteins or DNA. Either of these pathways may result in the development of an autoimmune condition/disease subsequent to toxicant exposure. As in many other situations, toxicant insult does not by itself necessarily initiate a negative result. Rather, a normal physiological response is distorted by the presence of toxicant so that a pathological response ensues.
The attached illustration covers these routes to autoimmunity plus situations where lymphoid cells expressing autoimmune immunoglobulins or T cell receptors evade elimination by the normal route of apoptosis:
The increasing use of
molecular biological tools such as protein expression arrays and PCR
will undoubtedly expand out
knowledge of normal immune regulation as well as provide important
biomarkers for evaluation of the presence of toxic
insults to the immune system.
Discussion Questions: