I urge everyone to
exert some extra effort to interact with
fellow students when responding to Discussion topics or questions to
minimize repetition in any written responses and the initiation of
discussion. Better would be discussion that includes peer
evaluation/correction and extension of initial ideas. Try to
avoid getting off on tangents or making obvious error in interpretation
by not asking for clarification or by not fully reading the questions
or textual materials.
Note, I do not have an absolute knowledge of every topic I raise. That's precisely where all of you come in. Each of you has a background knowledge that is somewhat different from the others and from me. Each of those backgrounds has strengths that should be built upon and shared. Do look at or think about what other people have said in their responses. Do check out the links students may have included in written responses or noted in their oral discussions (many are very good, but some could be questioned). Don't parrot the book, other websites, or other students. If you agree, say so. If not, or if you have questions, raise them. While there must be some repetition of material to answer questions, each student does not need to reiterate everything others have said. That's part of why it is good to jump in early to help set the tone of the Discussion or provide its basic elements. It's fine to agree if you see what you were going to write or contribute orally. But make sure you add something as well. I'll try to continue posting about two questions per session until the last couple of sessions. While there will be some objective components in these questions, there will be plenty to conjecture on or disagree with. I look forward to all of your responses.
Biochemistry of Toxicant Metabolism
Besides Casarett
& Doull, there is another resource text that is
commonly used in this arena: Goodman & Gilman's Pharmacological
Basis of Therapeutics, Pergamon Press. (The 8th edition
was edited by Gilman, Rall, Nies & Taylor and published in 1990, so
this text should be in about the 12th edition by now.)
Both
texts spend considerable time detailing information on the absorption,
metabolism, action, and excretion of chemicals. C&D focus on
toxicants
while G&G focus on clinical compounds. Both sources include
substantial
detail on chemistry, biochemistry, and enzymology. The newer
texts also
put substantial emphasis on genetics and genetic diversity as it
impacts
chemical metabolism. Indeed, this latter theme is very striking
in C&D
Chapter 6. There's also a recent popular book out that deals with much
of this topic, Why Some Like it Hot
which discusses the interaction between genetic diversity, nutrient
intake, disease susceptibility, and development of particular cuisines.
Among the mountain of material covered under Toxicant Metabolism a few items stand out.
First, is that xenobiotic metabolism is a variant or outgrowth of the metabolism of endogenous substances. These processes have not evolved simply to handle man-made substances. Rather, they have been exploited by the organism to cope with all chemicals, endogenous, natural environmental, or synthetic to which an organism is exposed. Likewise, the ability of drugs or synthetic chemicals to induce or alter the genetic transcription of metabolic enzymes is due to the ability of those drugs or synthetics to occupy naturally occurring receptors or to modify the function of naturally occurring transcription factors. These processes are not new to modern day organisms. They have been commandeered by newly available chemical forms. Even drug resistance proteins and the development of pesticide resistant organisms are the result of selection pressures making use of an existing genetic substrate. I am unaware of good evidence for the production of proteins with totally new enzymatic functions being generated without the use of transgenic techniques in organisms that did not previously possess near molecular relatives or closely related genes. Evolution clearly, but revolution? Probably not, since 50 or 300 years is too short on an evolutionary time scale.
Second, the metabolic systems are generally arranged in two segments. Phase I normally involves the oxidation, hydroxylation, hydrolysis, and/or reduction of parent compounds. While this frequently makes lipophilic compounds less lipophilic, the change is often rather minimal. It does, however, introduce functional groups that can be acted upon by Phase II enzymes that usually conjugate compounds to hydrophilic groups such as glucuronic acid, sulfonic acid, amino acids or glutathione. These latter groups frequently allow the conjugates to be filtered into urine in the kidney glomerulus but also help prevent them from being reabsorbed in the kidney or gut.
Note that some compounds will be eliminated without being metabolized, e.g., ethyl ether. Others may be eliminated immediately after phase I, often via the bile and feces, without the action of phase II enzymes, e.g., some hydroxylated steroid metabolites. Yet other compounds may interact directly with phase II enzymes without being processed by phase I enzymes.
Third, there is considerable redundancy in the capacity to metabolize many compounds. Not only are multiple forms of P450 (CYP gene products) often available, but some of the same reactions can occur via the actions of other enzymes, e.g., flavin monooxygenases. This redundancy means it is very difficult to totally inhibit a chemical's metabolism by the use of specific enzyme inhibitors or to explore the impacts on metabolism using transgenic animals (knockout mice may lack the target enzyme but simply use another pathway to metabolize the chemical of interest).
Fourth, there is tremendous variability among tissues, organs, and organisms in their capacity to act on xenobiotics. Some of this is due to tissue-specific expression of enzymes or transcripts, some is due to environmental exposures to modulators of enzyme expression. In addition to genetic composition differences among individuals, there is variation by gender, developmental stage, and nutritional status. And, because many of the genes involved can be expressed as alternative transcripts that may differ in their enzymatic kinetics and these can condition the expression of other enzymes in either phase I or phase II, there is a certain stochastic element in the expression of xenobiotic degradative enzymes. Note how this almost hypervariability complicates the task of toxicity testing, particularly in discerning effects at low dose levels. Note also how the differences of enzyme expression in tissue and enzyme type that exist among species also cloud the task of extrapolating test results obtained in one species to regulation of compound exposure in other species.
Fifth, not all metabolism leads to detoxication. In fact, there are many instances in which activation or toxication occurs as a result of the actions of phase I enzymes. Indeed, many drugs or toxins act only after phase I modification, e.g, parathion can only act as a nerve toxin in insects after conversion to paraoxon. Moreover, when toxication reactions exist, they often precede other reactions that are detoxifying. The balance of these reactions determines where a particular dose lies on the dose-response curve. If toxication saturates at low doses, detoxication may be able to keep up and minimize the impact of the toxicant produced in the first stage. If toxication saturates at higher doses than detoxication, detoxication cannot keep up and the toxic effects will manifest themselves until exposure is discontinued, toxication no longer continues, and detoxication catches up and reduces toxicant levels below minimal effect levels.
As a corollary here, the complexity of the toxicant activation/inactivation paths may include multiple tissues with activation in one (e.g., liver) and inactivation in another (e.g., bladder). Such pathways cannot be replicated in simple in vitro models. They force us to retain animal testing models. Moreover, they also mean that the location of toxic insults may also mimic the location of particular enzymes (toxifying enzymes if the toxic product moves slowly from the toxication tissue, i.e., the rate of toxification is greater than the rate of removal, or detoxifying enzymes if the toxic product accumulates in the site of detoxification, i.e., the rate of detoxification is less than the rate of production).
Sixth, metabolism is often stereoselective. This should not be surprising in a process making use of enzymes. These macromolecules and receptors are made up of L-amino acids and normally exhibit stereoselectivity for endogenous substrates or ligands. Thus, unless the intermediate formed during the biochemical reaction is symetrical and interacts directly with a small molecule like water that can enter the enzymatic reactive site, there is no reason why we should not expect the products of xenobiotic metabolism to be sterically selected.
Just how important hepatic, first pass, metabolism can be is demonstrated by the classic experiment (illustrated on pp 752-753 of C&D) of transplanting ovarian tissue to the capsule of the spleen while removing any additional gonadal tissue. The 17beta-estradiol made by the ovary passes via the portal vasculature directly to the liver where it can be very rapidly conjugated to glucuronic acid or sulfate or further metabolized to estrone and its similar conjugates. The conjugates and estrone are much less potent as estrogens than 17beta-estradiol, they do not bind as well, if at all, to the estrogen receptors in target tissues including the brain. As a result, the normal negative feedback loop controlling gonadotropin (LH and FSH) production by the gonadotropes of the anterior pituitary is broken and LH and FSH levels rise dramatically. But the gonadotropins can still act on the transplanted ovary. In doing so they stimulate tissue growth and steroid output. When continued chronically, this results in neoplastic growth of the ovary. Interestingly, a related situation arises in cases of genetic absence of the androgen receptor (testicular feminization syndrome, tfm). Here a genetic defect in the androgen receptor breaks the feedback loop by making the hypothalamus and gonadotropes insensitive to circulating androgen. The chronic stimulation by high LH and FSH often results in tumors unless the gonads are surgically removed. Note that androgen metabolism includes more alternative products than estradiol metabolism. As a result, transplanting the testis to the spleen would not have the same impact seen with the ovary. The first pass through the liver would not eliminate all active androgens from circulation and would not cause as dramatic a rise in LH and FSH.
Enzymatic reaction mechanisms are also referenced in C&D without explanation: a Ping Pong Bi Bi mechanism is a two stage mechanism involving biomolecular interactions at each step and a relatively stable (isolatable) modified enzymatic intermediate:
R1X + EY => EX + YR1
EX + R2Y => EY + XR2
where Y (and possibly R2) is often a simple group like H+ and OH-. Reactions can also be Ping Pong (R1X + E => EX + R1'; R2 + EX => R2X + E), having a stable enzymatic intermediate but no group released from the enzyme site during the reaction; or concerted (R1X + E => R1 + X + E) where no stable intermediate occurs and all reactant products are released at the same time; or
sequential (R1X
+ E => R1' + EX => E + X) where
no stable intermediate occurs and the products are released
sequentially,
etc. These are covered in most good textbooks on biochemistry or
enzymology.
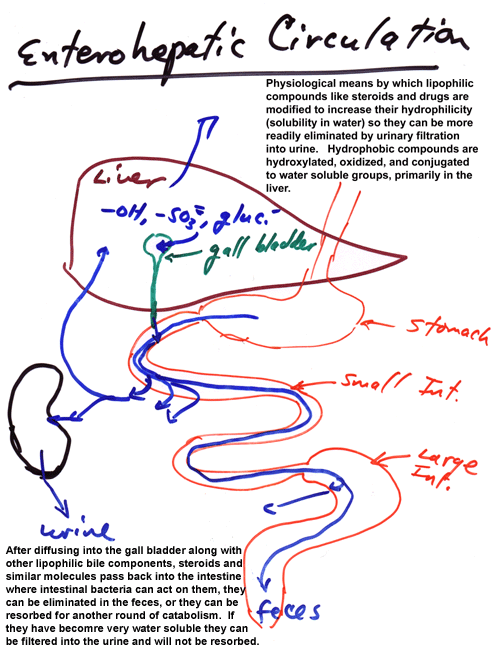
Enterohepatic Circulation
One element of xenobiotic metabolism that is mentioned briefly but not explored in any depth is enterohepatic circulation. This is an important concept for endogenous substrates such as steroids and plays an important role in tying together our earlier considerations of variations in gut physiology with the descriptions of xenobiotic metabolism covered by the text and related literature. Since many xenobiotics are lipophilic, they often end up being made slightly more hydrophilic by phase I metabolism (often in the liver). This does not make them sufficiently water soluble to be cleared in the kidney (often because they circulate in association with binding proteins that are not filtered by the kidney). Instead, they may diffuse or be transported into bile along with endogenous chemicals with similar physical chemical characteristics (e.g., cholic acid and its esters and taurine conjugates). These compounds, other bile elements, and pancreatic juices including pro-forms of enzymes that act on macromolecules or gastric breakdown products and lots of HCO3-2 are subsequently mixed with foodstuffs. As these are digested in the small intestine, monomers and small nutrients are absorbed along with water and mineral ions. Some lipids either diffuse through the intestinal wall or are transported as complexes by lipid transporters. Other lipids continue through the intestinal tract and may be acted upon by digestive bacteria, particularly in the large intestine (or cecum, if you have one), and transformed to additional metabolic products that may be more or less lipophilic than the original metabolites. Many of the lipophilic compounds are excreted as part of the feces, in free form, absorbed to particulates in fecal matter, or as components of bacteria eliminated during defecation. Those materials, especially lipophilic materials, resorbed during passage through the intestines reenter the circulation via passage into the mesenteric venous drainage or the mesenteric lymphatic system. Those that enter the mesenteric vein pass into the portal vein and thence reenter the liver. Those that enter the lymphatics may circulate again prior to re-exposure to hepatic enzymes. Passage through the liver a second time may result in further metabolism and disposal via the bile, thereby triggering a second round of enterohepatic circulation. Ultimately, compounds are usually conjugated by phase II enzymes to products that are sufficiently water soluble to circulate free in serum and undergo filtration and elimination by the kidney. There can be some retrieval of metabolites via anion transporters from the kidney tubules or lower intestine, adding to the enterohepatic circulation, but the basic system is as outlined.
Diagrammatically this is:
The details of this system and the extent of its impact on xenobiotic metabolism will vary with species, xenobiotic dose, diet (including soluble and insoluble fiber content), and prior modification of xenobiotic metabolism (induction or inhibition).
Note the role of the binding proteins in serum. These normally do not limit toxic actions because the ultimate targets are normally molecules with affinities one or more orders of magnitude higher than those of the carrier proteins. They do, however, make a difference in allowing glomerular filtration and in determining the bioavailable (free) concentration of xenobiotic or toxicant in circulation. This should be reflected in kinetic models but often is not since blood is considered a single compartment rather than a mixture of free and protein bound compound.
Toxicodynamics and Toxicokinetics
This refers to the movement of toxicants into, through, and out of biological systems. It considers not only route and concentration of dose, but also volume of distribution , bioavailability, perfusion and diffusion. The manner in which these play a role is often determined by which model of physiological distribution is most appropriate to the compound in question.
Note that Classical Models are data based and often require assumptions as to the uniformity of distribution or elimination of a toxicant. They tend to be simpler than Physiological Models but are less demanding to construct and evaluate as: "the appropriate physiological (e.g., blood flow rate, tissue volume) and biochemical (e.g., rate of biotransformation in a particular tissue) parameters [required for the Physiological Models] are often unknown or inexact, hampering meaningful physiologically based pharmacokinetic modeling." (Klaassen & Watkins, Casarett & Doull's Toxicology, Companion Handbook, p 134.) This latter restriction means that toxicologists must often run controls in parallel with their toxicant evaluations which will provide information on the basic processes involved. This adds to the value of toxicology studies but tends to slow them down. Note the Classical Models cannot predict tissue concentrations while well constructed Physiological Models can. The Physiological Model estimates are based on known or hypothesized biological processes, they require more basic data to compile, the math used to describe them is difficult (it requires calculus or iterative computer estimation of parameters), and the parameters involved are sometimes ill-defined.
The compartments used for the Physiological Models involve compartment size, flows into and out of these compartments, thermodynamic movement and partitioning between the compartments, and a consideration of the transport processes involved (e.g., perfusion versus diffusion limitations on transport between compartments).
Note the state assumptions for 1 compartment and 2 compartment systems.
1 compartment system: body is a homogeneous unit
2 compartment system: body is an inhomogeneous unit
In the 1 compartment system the clearance kinetics resemble a simple half-life plot as in radioactivity, where constant proportions of chemical are lost over unit increments of time. The plot of log tissue concentration versus time is a straight line with a single negative slope.
In the 2 compartment system the clearance kinetics have two or more phases and a plot of log tissue concentration versus time after dosage can have a rising, or loading, limb and a falling, or unloading, limb. The overall shape of the concentration versus time graph is actually the sum of these two limbs. Graphical analysis, then, resembles what is done to look at systems with high and low affinity binding sites as done in endocrinology via Scatchard plots, or what is done in enzymology to accommodate the existence of multiple enzyme forms having high and low enzymatic potential.
The comparments in these models are graphical or mathematical constructs that may or may not have a straight forward correlate within the biological system. An example of the different loading and unloading rates can be observed when choriogonadotropin (hCG) is labeled with two isotopes of radioiodine, one in each subunit. When 125Ialpha/131Ibeta hCG is administered to the female rat, plasma levels tend to be parallel for both isotopes with a rising and falling limb. But, there is a non-parallelism that is evident for the two isotopes as they are passed into the kidney. The data demonstrate the physical existence of at least two compartments for the processing of the two subunits of this glycoprotein molecule in this physiological system but it does not give details on where or how these compartments arise.
On the other hand Physiological Models attempt to simulate the rate controlling processes operative in vivo. Notice I said "rate controlling." Most of these models apply simplifying assumptions concerning what processes, e.g., flux across membranes, are not limiting and can therefore be dropped from inclusion in the equations. They also frequently assume steady state conditions apply to equilibria across membranes or other diffusional barriers or to enzyme reactions involved in metabolism. That is not to say they do not simulate in vivo conditions for model organisms very well. There are reasonable questions that can be raised for either type of model as to how well it can be extrapolated to other organisms given the variations in metabolic capacities mentioned earlier. Allometric scaling among taxa is an approximation in the best of circumstances and that is also true here.
Extrapolations across homeothermic/poikilothermic or across vertebrate/invertebrate lines may be particularly problematic for toxicokinetic models (especially Physiological Models) due to their heavy dependence on chemical partition coefficients. This is because such coefficients are sensitive to temperature. So while there may be no problem in making comparisons among mammals whose core body temperatures are maintained within a narrow range between about 35 and 40ºC, these coefficients will not hold for fish, reptilians, amphibians, or invertebrates. Even for mammals there may well be external temperature impacts for dermal absorption on the extremities.
Apparent distribution volume (Vd):
Although computed as (the initial bolus iv dose)/(the integrated area under the clearance curve) x (the elimination rate constant ß), the Vd is equivalent to the (actual distribution) x ((affinity within distribution sites) + (any metabolism in those sites)). As such, it may yield a calculated distribution volume that is greater than vascular (or even body) volume. In the simplest case, such as inulin, which is not metabolized and does not penetrate beyond the vascular space, solving the equation below for x:
(# of counts of administered radiolabeled inulin/x) = (measured counts/measured plasma volume)
gives the actual total vascular volume. For other compounds the distribution volume will the > or < the total vascular volume because the compound may be bound or sequestered and held out of circulation or it may be cleared and thus removed from circulation. Note that deposition of a toxicant into a storage deposit such as fat or bone mineral matrix prevents movement of the compound through other metabolic compartments or toward elimination. This "fixation" will greatly increase the apparent distribution volume because it decreases beta and increases the integrated area under the clearance curve.
A similar, related situation pertains when the compounds of interest can bind to serum binding proteins. Such proteins act to slow clearance and thereby expand the apparent distribution volume of the compound involved. Note the role of specialized transport systems in facilitating toxicant distribution are counterpoised to the impacts of plasma and intracellular/intercellular binding sites for serving as reservoirs that decrease bioavailable dose while allowing for bioaccumulation and/or prolonging exposure. Indeed, the consideration of bioavailability provided in C&D under toxicokinetics omits this very common but important point: to be equivalent to the bioavailability concept applied to endogenous chemicals and to many drugs the bioavailabilty factor designated F (= free) should strictly be limited to consideration only within target tissues and its computation should involve a dose adjustment factor reflecting the binding of the compounds to serum binding proteins. F as defined in C&D as the fraction of dose absorbed systemically, that is, extravascularly, is based on measurements of total serum or plasma content of compound. By contrast any measurement of free steroid or thyroid hormone is based on measurements of the concentration of hormone found in a small protein free compartment during equilibrium dialysis against a large volume of hormone containing serum. Or, equivalently, the amount of hormone capable of capture by a small amount of low affinity antibody for that ligand. In both hormonal measurements, the idea is to avoid disturbing any protein-bound hormone versus free hormone equilibrium in serum and to measure only that portion that is free in solution. Thus, as defined by C&D, F does not measure free xenobiotic. It measures absorption from vascular space only. When
F = (AUCpo/Dosepo) x (Doseiv/AUCiv)
where AUCpo, iv are the Area Under the Clearance curves integrated from initial exposure to complete clearance, the data to compute AUC are normally measurements of total compound in circulation. While this equation minimizes some of the distributional problem by using a ratio of AUC by an indirect route of systemic exposure to a direct route, it does not consider differences in loading of serum proteins that may occur by the two routes as a result of differences in saturation of this circulatory depot. Therefore F, as calculated, may approximate, but probably overstates, the amount of compound actually accessible to the organism.
The related issues of clearance and half-life obviously depend on Vd and clearance rate. If a compound has a very large apparent distribution volume or a slow clearance rate, it will have a long half-life, e.g., DDT which deposits in fat. Likewise, if the Vd is small, and clearance rapid, the half-life will be short, e.g., many bioactive peptides such as glucagon are cleared very rapidly by serum proteases which essentially limit their actions to neighboring cells and produce half-lives of less than 5 min.
Things change, however, when clearance and/or distribution systems become saturated. Parent compounds accumulate and biological effects become demonstrable, often for as long as it takes to clear the affected tissue. Consider which ranges are often being examined when studies are conducted of pharmaceutical drugs on the one hand or environmental pollutants on the other. In the case of pharmaceuticals, it is important to achieve and maintain an effective dosage for as long as necessary to induce a clinically beneficial response. This normally means that the system must be "loaded" to saturate degradative and non-target distributional systems as well as to potentially establish an internal deposit that may serve to prolong drug action. Failure to achieve this saturated state will limit the time of action of the drug or may even prevent the drug from attaining an effective target tissue concentration. When looking at pollutants or food contaminants, on the other hand, we are exploring toxicant effects that are frequently at the very low end of dose-response curves. These systems are often operating far from saturation and may well be readily analysed by very simple models such as the one or two compartment classical models.
Finally, please
notice that there are some
ambiguities in the molecular forms being tracked in several of the
toxicokinetic
models. It is entirely possible, if a specific measurement of the
intact
parent molecule is being used, e.g., via mass spectral analysis of a
specific
chromatographic fraction, to miss an important set of reactions
involving
a biologically labile conjugate. For example, if a compound is
conjugated
in the liver but is deconjugated in the intestine prior to
reabsorption,
the conjugate may not occur in the same chromatographic fraction as the
parent compound and it will be missed in the analysis. The reentering
compound
will simply contribute to the plasma pool and will not be
differentiated
as recycling compound. Likewise, use of too condensed a model, e.g.,
the
Classical Models, may also miss biologically important intermediates in
the distribution and degradation components of molecular dynamics. If
radiotracers
are being used it is also possible to miss important details because
the
radioactive tags do not by themselves differentiate among parent and
metabolite
forms. In addition, radiotags such as radioiodine or certain tritium
labels
can be easily lost early in metabolism. These then will fail to report
on the fate of major, potentially biologically important forms of the
xenobiotic
being evaluated.