Mammalian Toxicology, Session 2
Toxicant targets; Physiologic dose-response; C 2, 21; The role of intercellular chemical communication: hormone, receptor, transducer, effector; agonist, antagonist; Interconnections of transduction mechanisms; C 3; End of Add/Drop
Toxicant targets:Lethality: mortality, death (induced reasonably rapidly as an obvious consequence of exposure)
Morbidity: a decrease in functionality that may ultimately lead to a decrease in lifespan
Reduced reproductive capacity: decrease in reproductive output (this has a genetic carry-over to the next generation because there is a resulting change in the potential gene pool)
Decrease in specific functions or acuities (sensory: sight, hearing, taste, smell, touch; specific metabolic functions - food allergies; appearance - hair growth, acne): these decrease quality of life but are often nonlethal, do not decrease lifespan and do not alter the gene
Reduced asthetics or other quality of life issues: these are often issues that have little or no medical impacts except at the psychological or emotional levels
Reduced quality of the environment: this may impact any or all of the above items
Note that these range from severe immediate impacts on the individual, through prolonged or delayed impacts on the individual or the species, to subtle effects that may only alter survival or functions of individuals or species via alterations of behavior or mental vitality. Evaluating risks of toxic exposure then may encompass analysis of biological risks and exposure tests, potential medical or economic risks of exposure to or lack of exposure to the toxicant of interest, and possible impacts on the integrity or improvement of environmental quality. The overall picture may involve a variety of competing interests or benefits and definitely involves a cost/benefit analysis. Even at the individual level this is reflected in the statements that exist on drug and household chemical packaging. For medications there are not only directions for proper use, but indications of possible deleterious side effects. These are often unrelated to the drug target tissue of interest, e.g., antihistamines not only decrease swelling in nasal mucus membranes, they often suppress mental alertness and/or increase blood pressure. The detailed analysis of positive and negative drug impacts is subsumed in the science of pharmacology but has obvious overlap and relation to toxicology both in its tenets and methodologies but also in the subjects of study and the analyses used to evaluate study results.
Targets of toxic insult:
A chemical can act on an organism in two ways: directly and indirectly.
To act indirectly, it may disrupt the functioning of a cell or tissue that communicates with, regulates, or supports another cell or tissue. Thus, a disruptor of neural or pituitary control over adrenal function may generate a toxic response that is due to failure to produce proper amounts of the glucocortical steroids that are a main product of the adrenal cortex. So although the toxicant is acting in tissues in the head its manifested toxic response may appear in altered immune function (suppressed by high glucocorticoids) or in impaired growth (which requires the anabolic actions of glucocorticoids) of body structures. Obviously, teasing apart such indirect actions requires a keen knowledge of the normal regulatory circuitry within the body. The importance of background knowledge of endocrinology, neurobiology, and immunology to a toxicologist cannot be overstated.
To act directly, it usually has an impact on the cells and tissues that can be either general or specific. If it acts generally, it does so via a physical or chemical reaction that can be explained in generic terms: corrosivity - high acidity, highly caustic; chaotropism; detergency; solvency; chemically reactivity - forming adducts with broad classes of compounds like amines, alcohols, aldehydes, ketones, or alkenes that are present in many biological macromolecules; or generally proteolytic, lipolytic, or glycolytic - for example enzymes that might be found in bacterial or mycotic toxins or snake venoms. These actions can occur in nearly any or all exposed tissues or cells so the site of exposure will normally manifest the highest degree of toxic insult. Other toxic responses may also be induced in a secondary, or indirect, manner such as sensitization of the immune system via antibody production against chemically altered proteins or DNA in the originally exposed tissue, compromise of hearing if the tympanic membrane is disrupted by chemical or toxic actions.
The role of intercellular chemical communication: hormone, receptor, transducer, effector; agonist, antagonist:
If a toxin or toxicant is to act with some specificity as many, many do, it must interact with the cellular and tissue target in a specific manner. To do this it has to mimic or structurally resemble a molecule that is endogenous which binds to a particular location on or within a macromolecule in a cell or tissue. This might be an enzyme reactive site, an ion channel within a cell membrane, the receptor protein for a particular hormone in the membrane or nucleus of a cell, or a particular site on a DNA molecule. Note that all these locations have evolved shapes that allow them to act as conduits for receiving, transducing, or acting on chemical information passed to them from outside the cell or from other parts of the cell. That they have evolved these shapes suggests that they have done so in response to "normal" inputs, not extraordinary ones that may have been developed by synthetic chemists within the past 50 years. This means that such chemicals are indeed acting as structural mimics to endogenous or frequently encountered environmental chemical cues (see receptor antagonists, C&D p. 17). Moreover, there are finite numbers of these interaction or binding sites within cells or tissues. Thus, such interactions are saturable at high enough concentrations of the toxicant involved. Also, because there are finite numbers of these sites, toxin or toxicant interactions are normally in competition with the binding of endogenous chemicals (ligands). The relative strengths of the interactions of the binding site with the endogenous ligand and with the toxicant (the affinity constants for binding endogenous ligand or toxicant) will help determine the effectiveness of the toxicant in acting on the tissue. Along with the actual concentration of the binding site and the concentrations of the endogenous ligand and the toxicant, the saturation of the binding site by these two compounds can be computed based on simple binding kinetics: [BT] == [B][T]Kbt, [BE] == [B][E]Kbe, and BT <==> T + B + E <==> BE, where B is the Binding site, T is the toxicant, and E is the Endogenous molecule. Note that concentrations of B are often on the order of picomolar to micromolar as are the concentrations of E, that the dimensions of Kbe are often in the range of 108 to 1011 M-1 (=L/M), and that the resulting saturation for B sites is often below 50 or even 10%.
While this may describe the biophysics of the binding of a toxicant, the biochemical actions may range from strict competition with the endogenous ligand to allosteric (other site) modulation of the actions of the endogenous ligand via binding to locations on the target, binding, molecule that differ from the endogenous ligand (though they may overlap sterically). Toxicants that are termed "agonists" will not only mimic binding to a site overlapping that of the endogenous ligand, but they will trigger a similar cascade of intracellular events that is identical to, or strongly similar to that of the endogenous ligand. This usually occurs because they alter the shape of the B molecule in a way similar to E and cause it to allosterically interact with other molecules, transducers, that act to transduce the initial interaction into intracellular signals that the cell then converts via other macromolecules, usually protein effectors, into metabolic and/or physical reactions. On the other hand, toxicants, that simply block the E binding site but do not initiate the same sequence of events that E does, are termed "competitive antagonists." Agents that block the actions of E without competing directly for its binding site would be "noncompetitive antagonists" or "uncompetitive antagonists" depending on the physicochemical description of the kinetics of the interaction. These kinds of antagonists may mimic the product or transition state(s) of an enzyme or they may cause formation of a nonproductive form of enzyme complex, receptor-ligand complex, or DNA-ligand complex without necessarily blocking the binding of the endogenous ligand. In endocrinology and neurobiology we also encounter molecules that bind to receptors which trigger cascades of signals within cells that modulate or counter the actions of endogenous ligands acting via different receptors. Clearly these are a form of noncompetitive antagonists (or agonists) that does not fit the frame of binding to a single target molecule. Toxins may well act in a similar manner (see also functional antagonists in C&D p. 17).
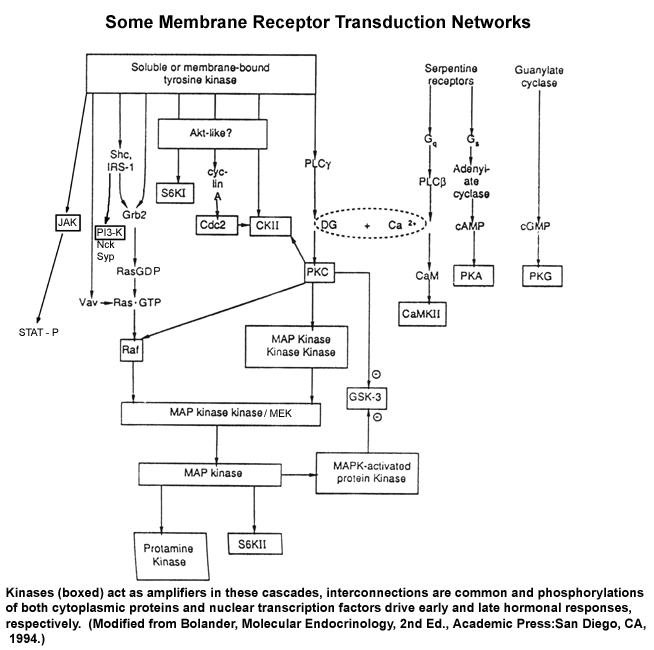
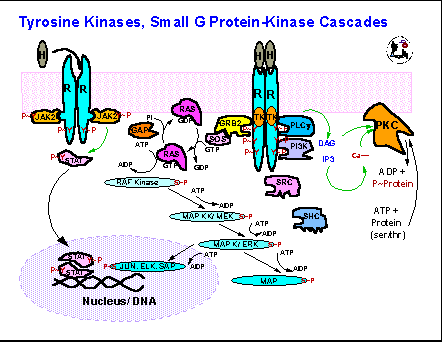
Interconnections of transduction mechanisms:
It is worth noting that many of the intracellular pathways linking extracellular chemical information inputs to cellular action have been defined (cf. any biochemistry, physiology, or endocrinology text). Moreover, as these have been more closely examined, it has also become clear that there are many instances in which these intracellular cascades cross and interact. That means that toxins may act via one or more of these pathways to activate or inhibit their normal functions and/or to change they way in which they respond to the usual extracellular signals. The following provide a couple of illustrations of this point:
In a few cases a toxicant acts not on a receptor but on a ligand or another toxicant directly, similar to the interaction between an antigen and an antibody. In these cases the first toxicant will be a chemical antagonist of the ligand or second toxicant.
Targets can also be general or specific. The generalized targets fit with nonspecific toxic insults as mentioned earlier: burns, sensitizations, tissue trauma resulting in necrosis (cell death normally induced by osmotic disruption of cell membranes caused by drastic alterations of cellular environment, low or high pH, solvent or detergent disruption of membranes, etc.).
Specific targets are often associated with specific organs, tissues, or cells. These in turn reflect special features of those organs, tissues, or cells often translating into similarities in surface proteins, lipids, or carbohydrates, or particular receptors or ion channels. Such entities will concentrate available toxicant molecules that bind to such molecular targets and will cause those tissues to act either as sites of toxic insult or as sites of toxicant metabolism, biotransformation and clearance. Note that sites of accumulation, for example fat depots accumulating lipophilic compounds via nonspecific physicochemical interactions between the toxicant and stored lipid molecules, will also concentrate toxicants. But they often lack target molecules of sufficiently high avidity for the toxicant molecules to bind them preferentially to those targets rather than the nonspecific lipid stores. When toxicants have no specific molecular targets, they often fail to elicit a specific toxic response.
Specific binding of toxicants also often reflects the status of another equilibrium in plasma and/or extracellular fluid, that is, the carrier or circulating binding protein bound form of the toxicant versus that found free in solution. Most lipophilic compounds and many other small ionic compounds tend to bind to sites on circulating carrier proteins which the body normally employs to distribute endogenous hormones and other molecules to their target tissues. Many of these carrier proteins are synthesized in the liver and often have binding affinity constants, Ka's, that are 10 to 1000 times lower than those of specific cellular receptors for a given hormone or endogenous molecule, i.e., 106 to 109 L/M. This allows such proteins to increase the volume to which endogenous ligands are distributed prior to loss of the ligand to metabolic, clearance, or simply receptor binding sites. They buffer the concentration of ligand in circulation and extend its biological "half-life" by decreasing its clearance from circulation. But these carriers often accept molecules of similar size or shape to their natural ligands. So toxicants can often be carried and distributed in this manner. Such carriers are rarely saturated with natural ligand beyond 50% of capacity, so there are often ample "spare" sites for toxicants to be carried even without interfering with natural ligand capacity. The binding reactions to such carriers are noncovalent and reversible. As free ligand increases in plasma due to endogenous synthesis or exogenous exposure, binding increases: B + L <==> BL, and the reaction proceeds to the right. As the complexes move to sites of lower free ligand, L, concentration, the reaction proceeds to the left generating more free ligand that can then bind to sites of higher affinity such as hormone receptors or specific toxicant binding sites. If free ligand is cleared by metabolic processes, the reaction also proceeds to the left decreasing the amount of complex remaining. Obviously, if enough carrier protein exists to tie up the majority of a toxicant until it is cleared, the biologically available concentration of the free toxicant many never exceed the minimum needed to generate a toxic insult. However, if metabolic or genetic alterations greatly diminish carrier binding capacity, a toxicant dose that may be readily tolerated by a normal individual may severely compromise the metabolically or genetically altered individual. Finally, note that many ligands can bind to several types of carrier proteins. Albumin acts as a remarkable "sink" for many lipophilic and ionic compounds but subclasses exist for many hormones, vitamins, and lipids (cf. C&D, p. 119-121.)
Selective Toxicity:
There are differing forms of toxicity. Acute lethality, effects on specific organs (that may or may not be potentially lethal), chronic, or subchronic effects (again that may or may not be ultimately lethal). In the case of chronic responses, there is historically a considerable emphasis on carcinogenesis as the endpoint of most concern. But what about neural degeneration, arthritic conditions, vascular rigidity, loss of muscle mass, loss of bone mass, or decline in immune function?
Note the definitions of lowest observable adverse effect level, LOAEL, and no observable adverse effect level, NOAEL. Note their dependencies on abilities to measure the chemical and the response within the biological system.
Physiologic Dose-Response:
While we looked at a few of the characteristics of the dose-response curve last time, there are more items to cover.
Definition of a toxicant and assumptions in dose response:
The empirical definition of a toxicant can be constructively viewed as a parallel phrasing of Koch's 1890 postulates regarding the empirical definition of a pathogen ( http://alan.kennedy.name/crohns/primer/koch.htm http://www.sci.wsu.edu/bio/micro310koch.html )